Definicja hipercholesterolemii rodzinnej (FH):
- choroba genetyczna monogenowa dziedziczona w sposób autosomalny dominujący
- prawie 100% penetracja wg praw Mendla - połowa dzieci pacjenta z heterozygotyczną postacią FH jest dotknięta chorobą
- charakteryzuje się wysokim poziomem LDL cholesterolu przy zwykle prawidłowych poziomach pozostałych frakcji lipidowych
- spowodowana jest mutacją genu receptora LDL, APOB-100 lub PCSK9
- mutacja genu receptora LDL powoduje brak lub zmniejszenie liczby receptorów LDL
- brak receptorów LDL uniemożliwia transport cholesterolu LDL do hepatocytów i jest przyczyną gromadzenia się w osoczu dużych ilości cholesterolu LDL
- nadmiar cholesterolu LDL gromadzi się w ścianach tętnic prowadząc stopniowo do powstania blaszki miażdżycowej
- proces miażdżycowy jest przyspieszony i skutkuje przedwczesną chorobą niedokrwienną serca.
Ocena ekspozycji na LDL cholesterol u pacjentów z hipercholesterolemią rodzinną
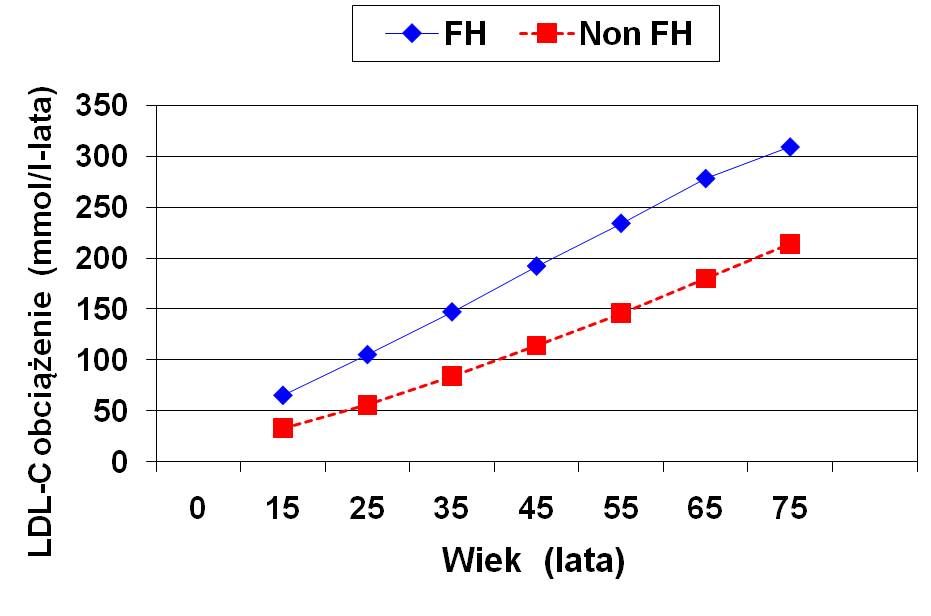
Do 45 roku życia heterozygota z hipercholesterolemią rodzinną doznaje ekspozycji na LDL cholesterol podobnej jak 70 latek bez hipercholesterolemii rodzinnej!!!
Epidemiologia hipercholesterolemii rodzinnej:
- ok. 30 milionów chorych z hipercholesterolemią rodzinną na świeci
- 190000 chorych w Polsce - odsetek chorych świadomych choroby jest znikomy
- postać heterozygotyczna występuje od 1 na 200 urodzeń
- postać homozygotyczna występuje 1 na 300 000 urodzeń.
Choroba wieńcowa w hipercholesterolemii rodzinnej:
- objawy choroby wieńcowej pojawiają się u ponad 50% pacjentów przed 55 rokiem życia
- wśród heterozygot przed 50 rokiem życia ryzyko zawału serca wynosi około 50%
- natężenie objawów zależy od stylu życia i dodatkowych czynników ryzyka.
Rozpoznanie kliniczne hipercholesterolemii rodzinnej:
- wywiad rodzinny (przedwczesna choroba wieńcowa)
- badanie fizykalne
- badania dodatkowe
- podwyższone stężenie całkowitego cholesterolu całkowitego i cholesterolu LDL
- wykluczenie przyczyn hipercholesterolemii wtórnej (zespołu nefrytycznego, cukrzycy, niedoczynności tarczycy, chorób wątroby, ciąży, farmakoterapii progestagenami, sterydami anabolicznymi czy glikokortykosteroidami).
Diagnostyka hipercholesterolemii rodzinnej
Postać homozygotyczna hipercholesterolemii rodzinnej:
- nie stwierdza się aktywności receptora LDL
- stężenie cholesterolu całkowitego zazwyczaj wzrasta 4-6-krotnie (600-1000 mg/dl), a LDL cholesterolu średnio 5-krotnie
- kępki żółte w skórze (Cutaneous xanthomas) powstają w ciągu kilku pierwszych miesięcy lub lat życia
- kępki żółte w ścięgnach (Tendon xantothomas) powstają nieco później i przyjmują postać guzowatą
- uogólniona miażdżyca rozwija się już w dzieciństwie i obejmuje tętnice wieńcowe, szyjne, biodrowe, udowe oraz aortę, a także może obejmować zastawkę aortalną
- choroba wieńcowa pojawia się zwykle około 10 roku życia
- choroba wieńcowa prowadzi do zawału serca przed 20 rokiem życia.
Postać heterozygotyczna hipercholesterolemii rodzinnej:
- podwyższony poziom cholesterolu można wykryć zaraz po urodzeniu lub nieco później
- wśród wielu osób dorosłych stężenie cholesterolu całkowitego i LDL cholesterolu wzrasta 2-3-krotnie
- poziom cholesterolu całkowitego osiąga najczęściej wartość 290-500 mg/dl zwykle bez towarzyszącego wzrost poziomu trój glicerydów
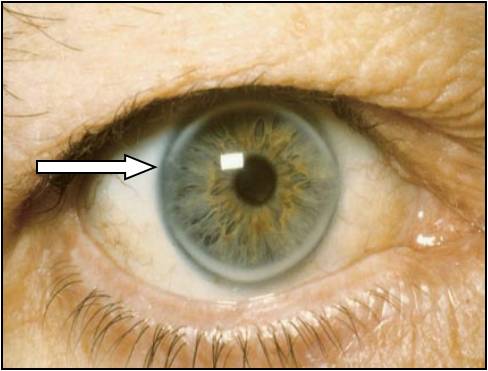
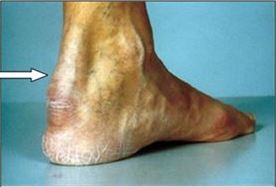
- między 25-30 rokiem życia pojawiają się żółtaki ścięgien (Tendon xantothomas), a także pierścień rogówkowy
- żółtaki ścięgien występują u około 75% chorych z postacią heterozygotyczną hipercholesterolemii rodzinnej
- żółtaki ścięgien są objawem patognomonicznym dla hipercholesterolemii rodzinnej
- pierścień rogówkowy występuje u 50% pacjentów poniżej 50 roku życia
Pierścień rogówkowy
Żółtaki ścięgien (Tendinous xanthomas)
- ruchome guzki ścięgien, więzadeł, powięzi i okostnej dłoni, palców, łokci, kolan i pięt
- najczęściej w ścięgnach Achillesa, w ścięgnach prostowników palców dłoni.
- mogą stymulować proces zapalny w ścięgnach, krwawienia i wysięk do tkanek
Źółtaki ścięgna Achillesa
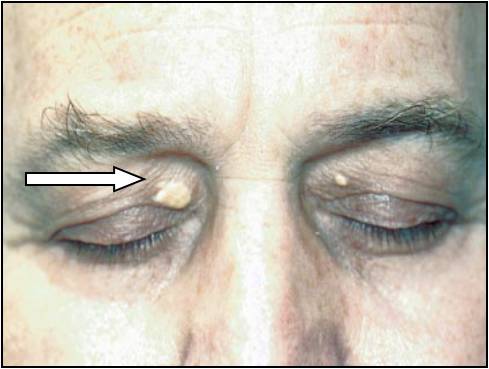
Żółtaki powiek (Xanthelasma)
- żółtawe plamki lub guzki w tkance podskórnej około oczodołowej
- nie są patognomoniczne dla hipercholesterolemii rodzinnej
Żółtaki powiek
Rozpoznanie kliniczne hipercholesterolemii rodzinnej- skala punktowa WHO (The Dutch Lipid Clinic Network)
Kiedy rozważyć rozpoznanie hipercholesterolemii rodzinnej?
- poziom cholesterolu całkowitego ponad 310 mg/dl i/lub cholesterolu LDL ponad 190 mg/dl przy współistniejącym
prawidłowym poziomie trójglicerydów
- przedwczesna choroba sercowo-naczyniowa (kobiety < 60rż., mężczyźni < 55rż.)
- pozytywny wywiad rodzinny w kierunku przedwczesnej choroby sercowo-naczyniowej
Podłoże genetyczne oraz podstawy diagnostyki molekularnej hipercholesterolemii rodzinnej dziedziczonej w sposób autosomalny dominujący (ADH, ang. autosomal dominant hypercholesterolemia, OMIM #143890)
- mutacje genu receptora LDL (hipercholesterolemia, AD, typ IIA / FH)
- mutacja genu apolipoproteiny B-100 (hipercholesterolemia, AD, typ IIB / FDB)
- mutacja genu PCSK9 (hipercholesterolemia, AD, 3/FH3).
Diagnostyka molekularna hipercholesterolemii rodzinnej:
- badanie molekularne genów związanych z hipercholesterolemią niezbędne do postawienia ostatecznego rozpoznania
- jest złotym standardem rozpoznania
- niezbędna dla wczesnego wdrożenia leczenia celem prewencji chorób sercowo-naczyniowych
- wpływa na obniżenie chorobowości oraz śmiertelności związanej z miażdżycą naczyń, szczególnie wieńcowych
- umożliwia objęcie rodzin z hipercholesterolemią rodzinną poradnictwem genetycznym
- umożliwia szybką i wiarygodną identyfikację osób chorych wśród członków rodzin probantów (diagnostyka kaskadowa).
Dziedziczenie hipercholesterolemii rodzinnej:
- choroba jednogenowa
- dziedziczy się jako cecha dominująca
- geny odpowiedzialne za powstanie hipercholesterolemii zlokalizowane są na chromosomach autosomalnych
- cecha taka występuje u obu płci z taką samą częstością
- heterozygoty mają jeden allel z mutacją i jeden prawidłowy
- homozygoty mają nieprawidłowe oba allele
Poradnictwo genetyczne - określenie ryzyka wystąpienia hipercholesterolemii rodzinnej:
- dla danej pary rodziców jest stałe
- dotyczy każdej ciąży
- nie zmienia się w zależności od liczby posiadanego potomstwa, chorego lub zdrowego
- w przypadku heterozygoty prawdopodobieństwo przekazania uszkodzonego allelu potomstwu wynosi 50%
- w przypadku homozygoty prawdopodobieństwo przekazania uszkodzonego allelu potomstwu wynosi 100%
Postępowanie diagnostyczne w hipercholesterolemii rodzinnej:
- ocena kryteriów klinicznych rozpoznania hipercholesterolemii rodzinnej u probanta
- wykluczenie wtórnej hipercholesterolemii
- kryteria kliniczne hipercholesterolemii rodzinnej wg Dutch Lipid Network
- wykonanie badań genetycznych u probanta
- w przypadku wykrycia mutacji u probanta badania molekularne członków jego rodziny
- wczesna profilaktyka i leczenie probanta i członków rodziny.